
Human ECE1 Protein (His Tag)
ECE,RP3-329E20.1
- 100ug (NPP3819) Please inquiry
| Catalog Number | P10887-H07H |
|---|---|
| Organism Species | Human |
| Host | Human Cells |
| Synonyms | ECE,RP3-329E20.1 |
| Molecular Weight | The recombinant human ECE1 comprises 698 amino acids and has a predicted molecular mass of 80 kDa. As a result of glycosylation, rh ECE1 migrates as an approximately 125 kDa band in SDS-PAGE under reducing conditions. |
| predicted N | His |
| SDS-PAGE | 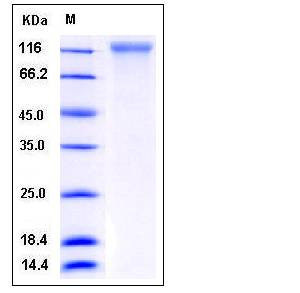 |
| Purity | > 95 % as determined by SDS-PAGE |
| Protein Construction | A DNA sequence encoding the extracellular domain of human ECE1 isoform B (P42892-1) (Gln 90-Trp 770) was expressed, with a polyhistidine tag at the N-terminus. |
| Bio-activity | |
| Research Area | Signaling |Signal Transduction |Growth Factor & Receptor |Hormones |
| Formulation | Lyophilized from sterile PBS, pH 7.4 1. Normally 5 % - 8 % trehalose and mannitol are added as protectants before lyophilization. Specific concentrations are included in the hardcopy of COA. |
| Background | Endothelin-converting enzyme 1, also known as ECE-1, is a single-pass type I I membrane protein which belongs to the peptidase M13 family. ECE-1 converts big endothelin-1 to endothelin-1. ECE-1 is a membrane metalloprotease that generates endothelin from its direct precursor big endothelin. Four isoforms of ECE-1 are produced from a single gene through the use of alternate promoters. These isoforms share the same extracellular catalytic domain and contain unique cytosolic tails, which results in their specific subcellular targeting.All isoforms of ECE-1 are expressed in umbilical vein endothelial cells, polynuclear neutrophils, fibroblasts, atrium cardiomyocytes and ventricles. Isoforms A, B and C of ECE-1 are also expressed in placenta, lung, heart, adrenal gland and phaeochromocytoma; isoforms A and C of ECE-1 in liver, testis and small intestine; isoform B, C and D of ECE-1 in endothelial cells and umbilical vein smooth muscle cells; isoforms C and D in saphenous vein cells, and isoform C in kidney. Defects in ECE1 are a cause of Hirschsprung disease, cardiac defects and autonomic dysfunction. It is a form of Hirschsprung disease with skip-lesions defects, craniofacial abnormalities and other dysmorphic features, and autonomic dysfunction. |
| Reference |